


I’ve spent the last few days writing an introduction for my first PhD paper on the practical issues of adding fossils to molecular phylogenies (full recipe here). This is my starting point: most people working in macroevolution agree that we should integrate fossils into modern phylogenetic trees. Of the many possible methods that are available, Ronquist’s total evidence method looks to be the most promising (however, some nice other ones also exist).
Recently Schrago et al. published a nice attempt to use this method on the Plathyrrini (New-World monkeys to you and me):
As a reminder, the aim of this total evidence method is to combine all of the available data: both molecular and morphological. Traditionally, analyses have treated each type of data separately; approaches which bring their own advantages and problems.
Let’s start with the molecules:
Opazo et al. published in 2006 a classical example of a molecular phylogenetics study. There are more recent, impressive phylogenetic studies (like Perelman et al. in 2011 and Springer et al. in 2012) on most of the primates and using more genetic data but I think Opazo is a better example of a traditional approach because it involves a tree with 17 taxa instead of more than 200.
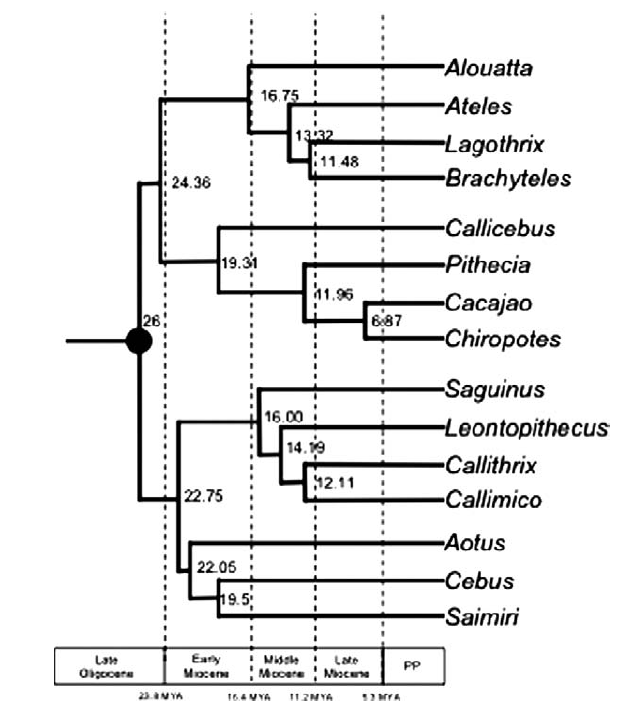
Two of the main advantages of this approach are the quantity of data involved (tens of thousands base pairs) and the methods of inferring the evolutionary history: molecular evolutionary models are easy to understand and easy to implement (each site has a finite number of states – A, C, G, T or nothing – and probabilistic models are good enough to infer the rate of changes from one state to the other). From a data perspective, another practical advantage is that, with modern NextGen sequencing, it’s really easy and fast to obtain a full genomic dataset. However, the main inconvenience from a macroevolutionary point of view is that molecular approaches don’t really take evidence from the fossil record into account. In the Opazo example, the only fossil used is Branisella, and the only useful information here is just its age (around 26 Ma) used to calibrate the time on the tree.
On the other hand, Kay et al. 2008 published an awesome study of the Platyrrhini history from a palaeontological point of view. They focused on 20 living taxa combined with 11 fossil species and using 268 morphological characters.
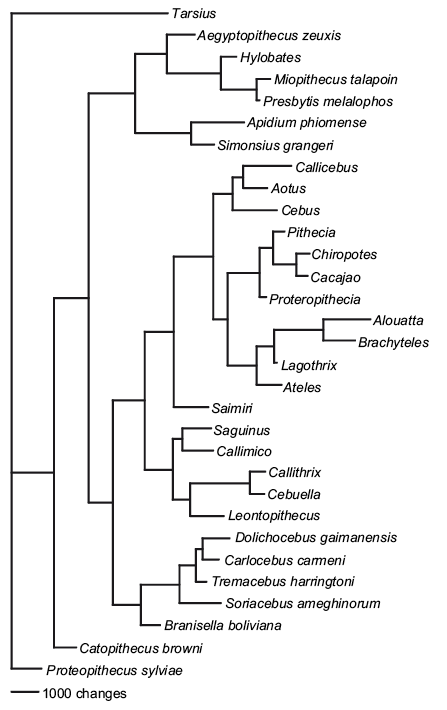
Again, there are both advantages and problems associated with this approach. Firstly, the number of characters used is pretty low; don’t get me wrong, 268 is really good for a morphological matrix, it’s just low compared to molecular data. Furthermore, the underlying evolutionary models used to build the phylogeny are hard to infer, the most common model is the Lewis 2001 Mk model where morphological characters are treated as if they “act like” molecular sites with no assumptions made about their states or rates of change (this method has been criticized but it’s still our best way to infer morphological evolution). Another solution, which is also commonly used, is to infer nothing but instead just use a maximum parsimony approach: find the tree which explains observed phenotypic evolution with the fewest number of evolutionary steps (characters changing from one state to another on a particular node within the tree). However, compared to a purely molecular approach, the advantages of Kay’s tree are clear from a macroevolutionary point of view: this tree includes full information from the morphologies of both living and fossil species!
Now hopefully you can see where I’m coming from in wanting to use the total evidence method? It’s clear from the empirical examples above that the problems associated with one approach are the advantages in the other. So let’s just combine them! And that’s what Schrago did in their work, they just mixed both data sets and re-ran the analysis (or, more precisely, they used Kay’s data set as it was but added new genomic data collected over the last seven years to Opazo’s data set). Here’s their result:
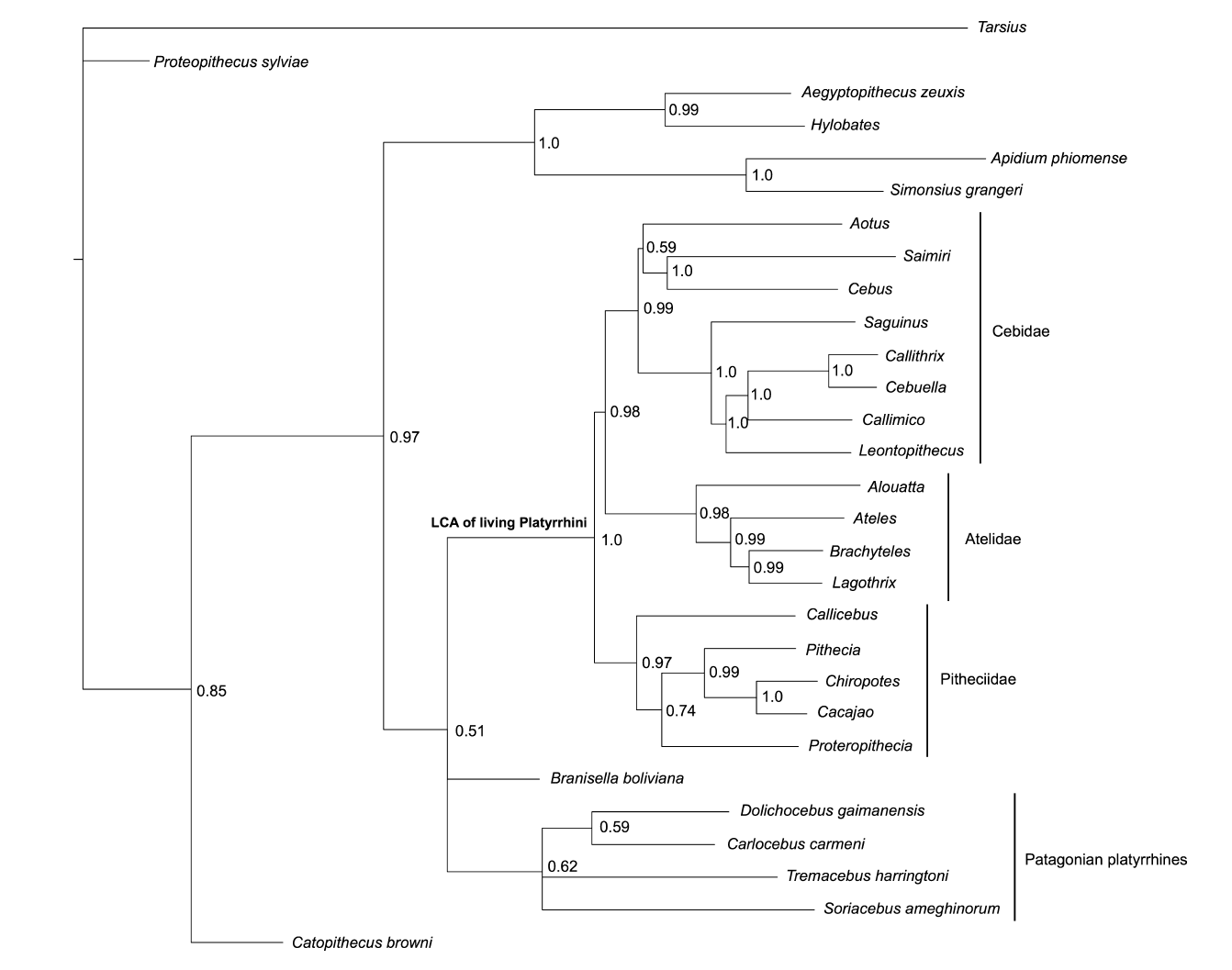
So here we have the advantage of both methods combined and this tree is far more user friendly for macroevolutionary studies; one can test evolutionary hypothesis through time using a more complete representation of the Platyrrhini evolutionary history. One major problem still remains though; the paucity of useful morphological data compared to the wealth of molecular data which is now available. Does that influence the tree’s topology somehow? Well, stay tuned, my simulations are running…
Author: Thomas Guillerme, guillert[at]tcd.ie, @TGuillerme
Photo credit: wikimedia commons